Seurat_Subset_Object
This project is maintained by brandonyph
suppressWarnings(library(Seurat))
## Registered S3 method overwritten by 'spatstat.geom':
## method from
## print.boxx cli
## Attaching SeuratObject
suppressWarnings(library(SeuratData))
## -- Installed datasets ------------------------------------- SeuratData v0.2.1 --
## v ifnb 3.1.0 v pbmc3k 3.1.4
## -------------------------------------- Key -------------------------------------
## v Dataset loaded successfully
## > Dataset built with a newer version of Seurat than installed
## (?) Unknown version of Seurat installed
pbmc <- suppressWarnings(LoadData("pbmc3k", type = "pbmc3k.final"))
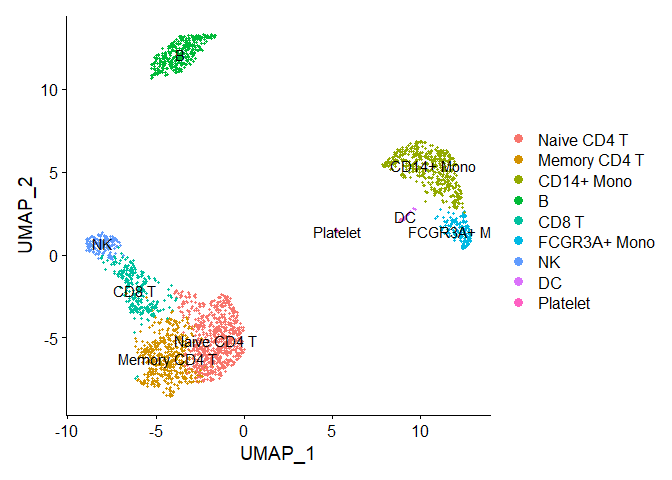
DimPlot(pbmc, reduction = "umap",label = TRUE)
# list options for groups to perform differential expression on
levels(pbmc)
## [1] "Naive CD4 T" "Memory CD4 T" "CD14+ Mono" "B" "CD8 T"
## [6] "FCGR3A+ Mono" "NK" "DC" "Platelet"
## [1] "Naive CD4 T" "Memory CD4 T" "CD14+ Mono" "B" "CD8 T"
## [6] "FCGR3A+ Mono" "NK" "DC" "Platelet"
Subseting based on the clustering
you can subset on mulitiple data wihtin the seurat object
-
Subset Seurat object based on identity class, also see ?SubsetData subset(x = pbmc, idents = “B cells”) subset(x = pbmc, idents = c(“CD4 T cells”, “CD8 T cells”), invert = TRUE)
-
Subset on the expression level of a gene/feature subset(x = pbmc, subset = MS4A1 > 3)
-
Subset on a combination of criteria subset(x = pbmc, subset = MS4A1 > 3 & PC1 > 5) subset(x = pbmc, subset = MS4A1 > 3, idents = “B cells”)
-
Subset on a value in the object meta data subset(x = pbmc, subset = orig.ident == “Replicate1”)
-
Downsample the number of cells per identity class subset(x = pbmc, downsample = 100)
CD14_pbmc <- subset(x = pbmc, idents = c("CD14+ Mono", "FCGR3A+ Mono"))
monocyte.de.markers.small <- FindMarkers(CD14_pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono")
# Find differentially expressed features between CD14+ and FCGR3A+ Monocytes
monocyte.de.markers <- FindMarkers(pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono")
# view results
head(monocyte.de.markers)
## p_val avg_log2FC pct.1 pct.2 p_val_adj
## FCGR3A 1.193617e-101 -3.776553 0.131 0.975 1.636926e-97
## LYZ 8.134552e-75 2.614275 1.000 0.988 1.115572e-70
## RHOC 4.479768e-68 -2.325013 0.162 0.864 6.143554e-64
## S100A8 7.471811e-65 3.766437 0.975 0.500 1.024684e-60
## S100A9 1.318422e-64 3.299060 0.996 0.870 1.808084e-60
## IFITM2 4.821669e-64 -2.085807 0.677 1.000 6.612437e-60
# Find differentially expressed features between CD14+ Monocytes and all other cells, only
# search for positive markers
monocyte.de.markers <- FindMarkers(pbmc, ident.1 = "CD14+ Mono", ident.2 = NULL, only.pos = TRUE)
# view results
head(monocyte.de.markers)
## p_val avg_log2FC pct.1 pct.2 p_val_adj
## S100A9 0.000000e+00 5.570063 0.996 0.215 0.000000e+00
## S100A8 0.000000e+00 5.477394 0.975 0.121 0.000000e+00
## FCN1 0.000000e+00 3.394219 0.952 0.151 0.000000e+00
## LGALS2 0.000000e+00 3.800484 0.908 0.059 0.000000e+00
## CD14 2.856582e-294 2.815626 0.667 0.028 3.917516e-290
## TYROBP 3.190467e-284 3.046798 0.994 0.265 4.375406e-280
Check for System Time (Speed of Execution)
system.time(FindMarkers(pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono", max.cells.per.ident = 500))
## user system elapsed
## 3.59 0.02 3.68
system.time(FindMarkers(CD14_pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono", max.cells.per.ident = 500))
## user system elapsed
## 3.76 0.01 3.94
system.time(FindMarkers(pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono", test.use = "DESeq2", max.cells.per.ident = 500))
## converting counts to integer mode
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## user system elapsed
## 66.94 1.61 71.28
system.time(FindMarkers(CD14_pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono", test.use = "DESeq2", max.cells.per.ident = 500))
## converting counts to integer mode
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## user system elapsed
## 61.94 0.63 68.89
time1 <- system.time(FindMarkers(pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono", test.use = "DESeq2", max.cells.per.ident = 500))
## converting counts to integer mode
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
time2 <- system.time(FindMarkers(CD14_pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono", test.use = "DESeq2", max.cells.per.ident = 500))
## converting counts to integer mode
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
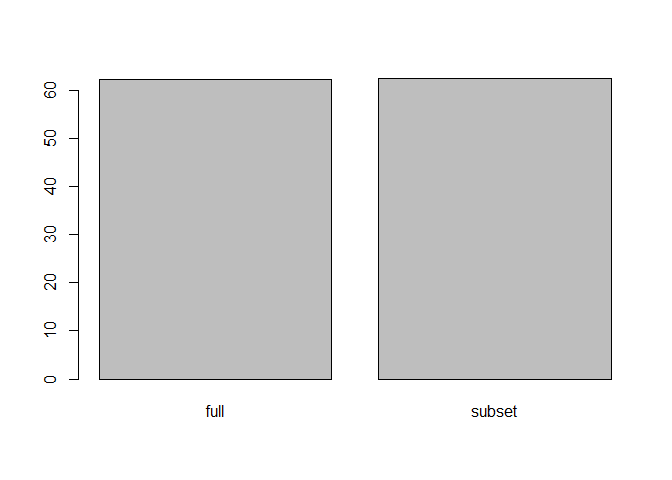
speed_df <- c(time1[3],time2[3])
names(speed_df) <- c("full","subset")
barplot(speed_df)